Multivariate Analysis of Transcript Splicing (MATS)
Xing Lab, Children's Hospital of Philadelphia
Updates
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Citation
Shen S., Park JW., Lu ZX., Lin L., Henry MD., Wu YN., Zhou Q., Xing Y.
rMATS: Robust and Flexible Detection of Differential Alternative Splicing from Replicate RNA-Seq Data.
PNAS, 111(51):E5593-601. doi: 10.1073/pnas.1419161111
Park JW., Tokheim C., Shen S., Xing Y. Identifying differential alternative splicing events from RNA sequencing data using RNASeq-MATS. Methods in Molecular Biology: Deep Sequencing Data Analysis, 2013;1038:171-179 doi: 10.1007/978-1-62703-514-9_10
Shen S., Park JW., Huang J., Dittmar KA., Lu ZX., Zhou Q., Carstens RP., Xing Y. MATS: A Bayesian Framework for Flexible Detection of Differential Alternative Splicing from RNA-Seq Data. Nucleic Acids Research, 2012;40(8):e61 doi: 10.1093/nar/gkr1291
Park JW., Tokheim C., Shen S., Xing Y. Identifying differential alternative splicing events from RNA sequencing data using RNASeq-MATS. Methods in Molecular Biology: Deep Sequencing Data Analysis, 2013;1038:171-179 doi: 10.1007/978-1-62703-514-9_10
Shen S., Park JW., Huang J., Dittmar KA., Lu ZX., Zhou Q., Carstens RP., Xing Y. MATS: A Bayesian Framework for Flexible Detection of Differential Alternative Splicing from RNA-Seq Data. Nucleic Acids Research, 2012;40(8):e61 doi: 10.1093/nar/gkr1291
About MATS
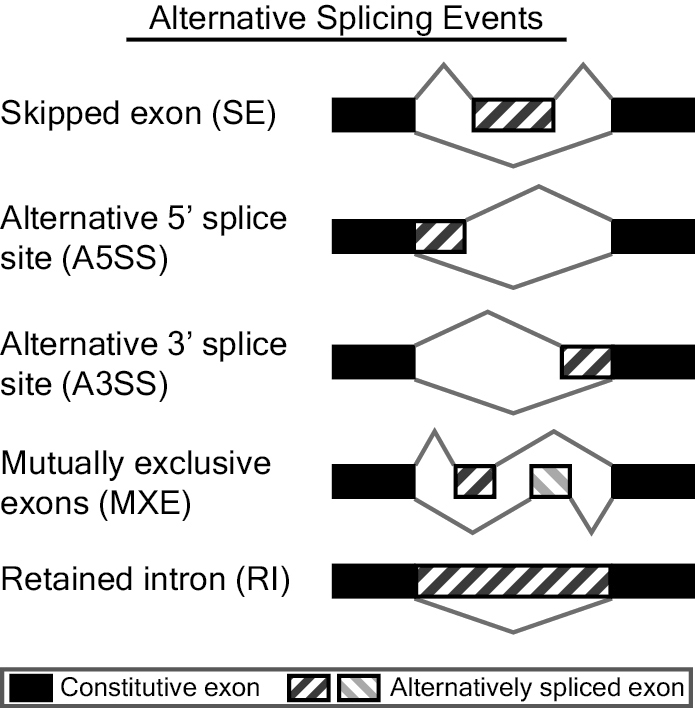
MATS is a computational tool to detect differential alternative splicing events from RNA-Seq data. The statistical model of MATS calculates the P-value and false discovery rate that the difference in the isoform ratio of a gene between two conditions exceeds a given user-defined threshold. From the RNA-Seq data, MATS can automatically detect and analyze alternative splicing events corresponding to all major types of alternative splicing patterns. MATS handles replicate RNA-Seq data from both paired and unpaired study design.
Software Download
|
|
|
|
Pre-requisites
|
Documentation
rMATS Companion
|
|
|
|
Questions/Comments
Have comments or questions about rMATS? Please post them on the rMATS Users Google Group.